

cheap fake rolex review absolutely has qualification to compare with the masterpiece of palace-level art..specific type is known as a rolex richard mille rm 030 replica includes.
Partnership with Avant-garde Materials Simulation
Solving crystal structures from Powder diffraction data
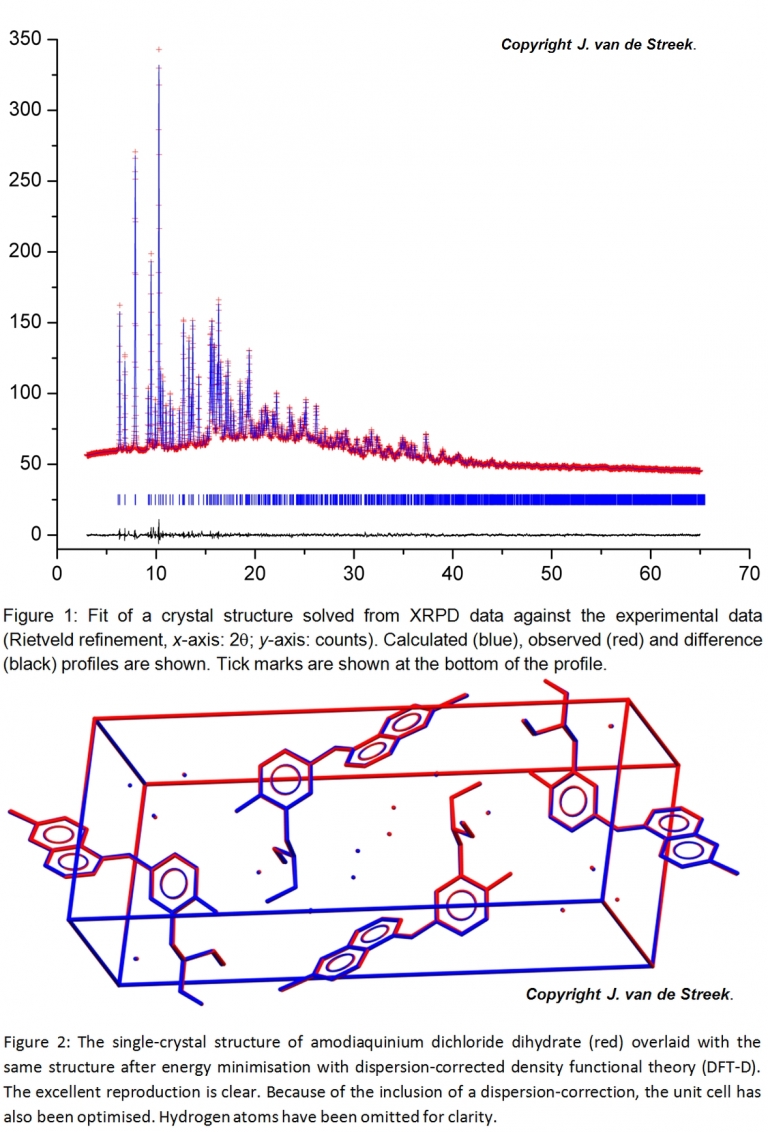
In many cases, molecular crystal structures can be determined directly from their powder diffraction patterns (Figure 1). This is especially appealing when SR-XRPD data is available, because the use of synchrotron radiation substantially reduces the peak widths, thereby resolving more peaks, and SR-XRPD patterns therefore ensure more accurate and more reliable crystal structures. Furthermore, a major bottleneck in crystal structure solution from XRPD is the determination of the unit cell (the indexing step) and this step is also greatly facilitated by the highly accurate peak positions as found in SR-XRPD data.
Therefore, in 2017, a partnership was built between Excelsus Structural Solutions and Avant-garde Materials Simulation, with the goal to provide customers with reliable crystal structures derived from high-resolution synchrotron-radiation X-ray powder diffraction patterns.
Avant-Garde Materials Simulation (AMS) is the world leader for Crystal Structure Prediction contract research and software (GRACE). Furthermore, scientists at AMS have been active in the field of structure determination from XRPD for more than 15 years, and between them they have solved over 50 molecular crystal structures from XRPD. Dr Marcus Neumann is the author of the program X-Cell for indexing of powder diffraction patterns [1] and Dr Jacco van de Streek is one of the authors of the DASH program [2].

Complementarity of X-ray powder diffraction and dispersion-corrected density functional theory
The structural information that is lost in a powder diffraction pattern due to peak overlap can be recovered by adding external information in the form of energy minimizations with dispersion-corrected density functional theory (DFT-D). DFT-D is a quantum-mechanical method that is able to reproduce organic crystal structures, even of large molecules, with unprecedented accuracy (see Figure 2); DFT-D this is the quantum-mechanical that proved so successful in the last three crystal structure prediction blind tests [3]. XRPD and DFT-D offer two complementary views of the crystal structure: a structure-based view and an energy-based view. When the two techniques agree on the same structure, the crystal structure is almost certainly correct. The DFT-D method has been quantitatively validated against more than 400 organic crystal structures [4,5] and has been applied to dozens of published organic crystal structures determined from XRPD.
[1] M. A. Neumann (2003). "X-Cell: a novel indexing algorithm for routine tasks and difficult cases" J. Appl. Cryst. 36, 356-365.
[2] W. I. F. David, K. Shankland, J. van de Streek, E. Pidcock, W. D. S. Motherwell & J. C. Cole (2006). "DASH: A Program for Crystal Structure Determination from Powder Diffraction Data" J. Appl. Cryst. 39, 910-915.
[3] Reilly, A. et al. Report on the sixth blind test of organic crystal structure prediction methods Acta Crystallographica. Section B: Structural Science 72: 439-459 (2016).
[4] J. van de Streek & M. A. Neumann (2010). "Validation of Experimental Molecular Crystal Structures with Dispersion-corrected Density Functional Theory Calculations" Acta Cryst. B66, 544-558.
[5] J. van de Streek & M. A. Neumann (2014). "Validation of Molecular Crystal Structures from Powder Diffraction Data with DFT-D" Acta Cryst. B70, 1020-1032.
Avant-garde Materials Simulation (Deutschland) GmbH
Rosa-Luxemburg-Str. 14 | D-79100 Freiburg, GERMANY.
Phone: +49 761 58997490
Fax: +49 761 58997495
Email: info@avmatsim.eu

